The main purpose of P-value Analysis was to use the Tooth Growth dataset. I wanted to showcase various R packages such as ggpubr and dplyr that use default methods such “wilcos.test”,“t.test”" etc. I intend to add more details of this analysis later. I wanted to demonstrate the working code here first.
if(!require(devtools)) install.packages("devtools")## Loading required package: devtools## Warning: package 'devtools' was built under R version 3.3.3devtools::install_github("kassambara/ggpubr")## Skipping install of 'ggpubr' from a github remote, the SHA1 (cb0f308d) has not changed since last install.
## Use `force = TRUE` to force installation#install.packages("ggpubr")
#install.packages("dplyr")
if (!require("ggpubr")) {
install.packages("ggpubr", repos="http://cran.rstudio.com/")
library("ggpubr")
}## Loading required package: ggpubr## Loading required package: ggplot2## Warning: package 'ggplot2' was built under R version 3.3.3## Loading required package: magrittr## Warning: Installed Rcpp (0.12.9) different from Rcpp used to build dplyr (0.12.11).
## Please reinstall dplyr to avoid random crashes or undefined behavior.if (!require("dplyr")) {
install.packages("dplyr", repos="http://cran.rstudio.com/")
library("dplyr")
}## Loading required package: dplyr## Warning: package 'dplyr' was built under R version 3.3.3##
## Attaching package: 'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union#library(dplyr)
#library(ggpubr)
#library(ggplot2)
data("ToothGrowth")
head(ToothGrowth)## len supp dose
## 1 4.2 VC 0.5
## 2 11.5 VC 0.5
## 3 7.3 VC 0.5
## 4 5.8 VC 0.5
## 5 6.4 VC 0.5
## 6 10.0 VC 0.5p <- ggboxplot(ToothGrowth, x = "supp", y = "len",
color = "supp", palette = "jco",
add = "jitter")
# Add p-value
p + stat_compare_means()## Warning: package 'bindrcpp' was built under R version 3.3.3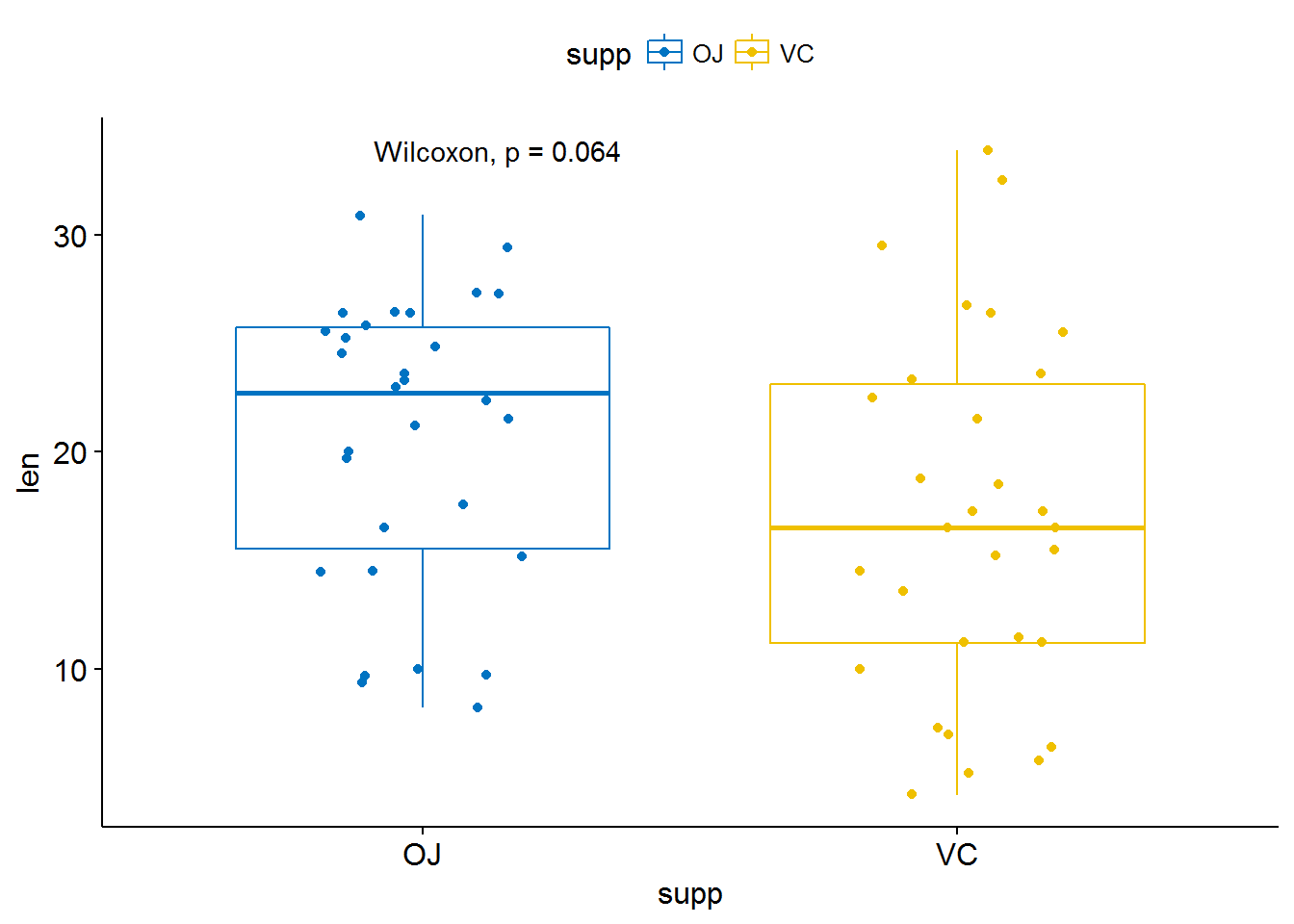
# Change method
p + stat_compare_means(method = "t.test")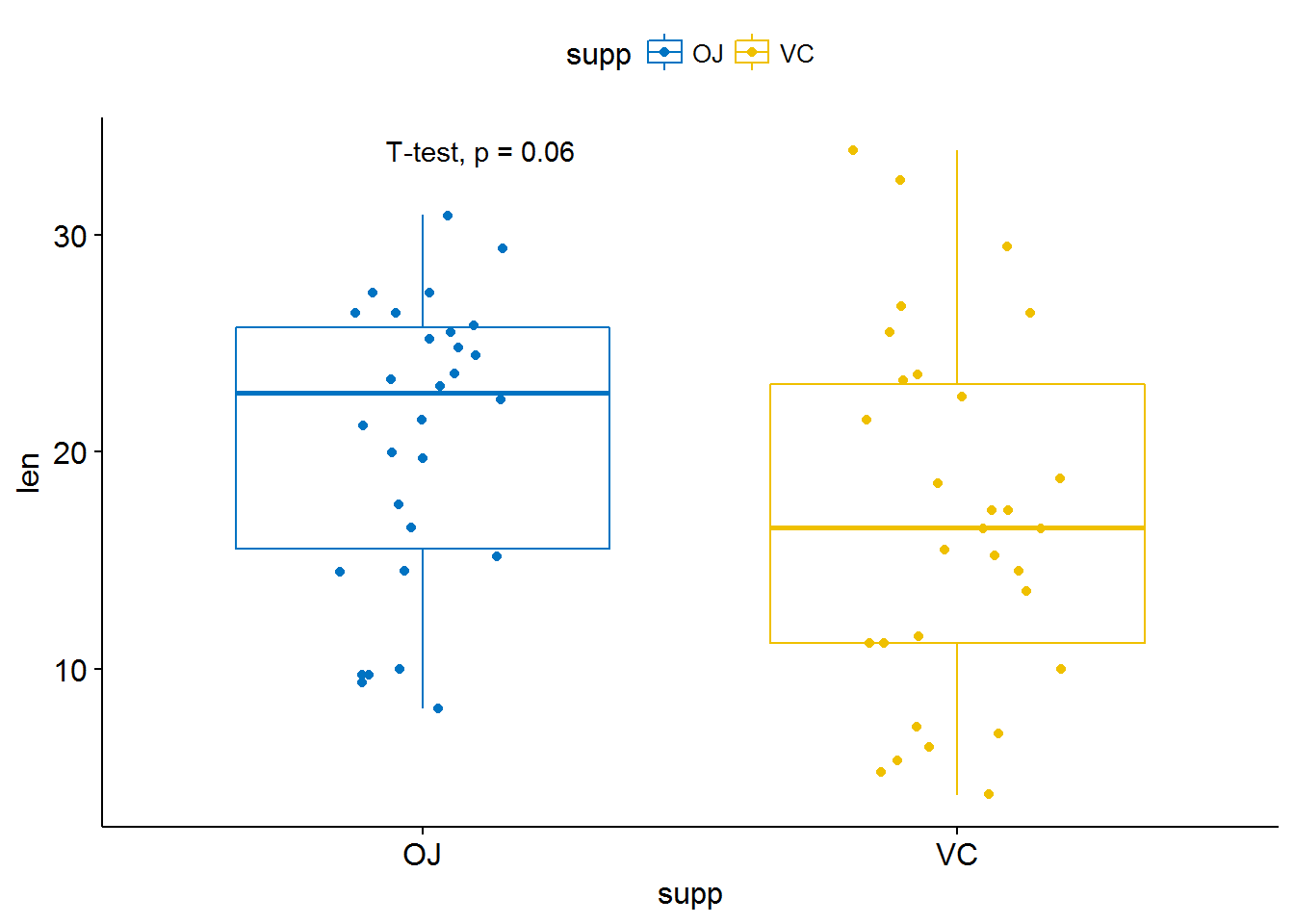
# Global test
compare_means(len ~ dose, data = ToothGrowth, method = "anova")## # A tibble: 1 x 6
## .y. p p.adj p.format p.signif method
## <chr> <dbl> <dbl> <chr> <chr> <chr>
## 1 len 9.532727e-16 9.532727e-16 9.5e-16 **** Anova# # Default method = "kruskal.test" for multiple groups
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
stat_compare_means()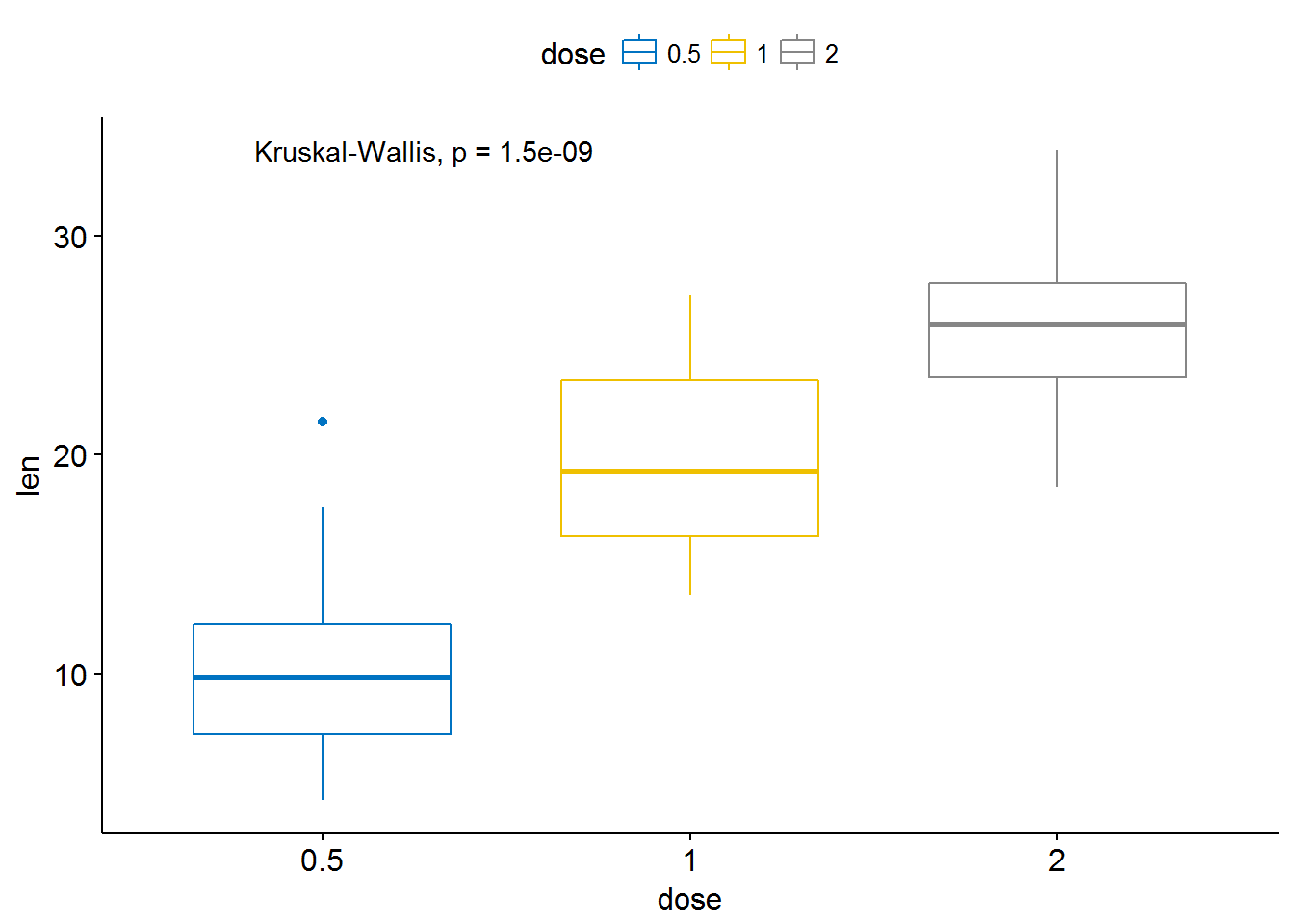
#
# # Change method to anova
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
stat_compare_means(method = "anova")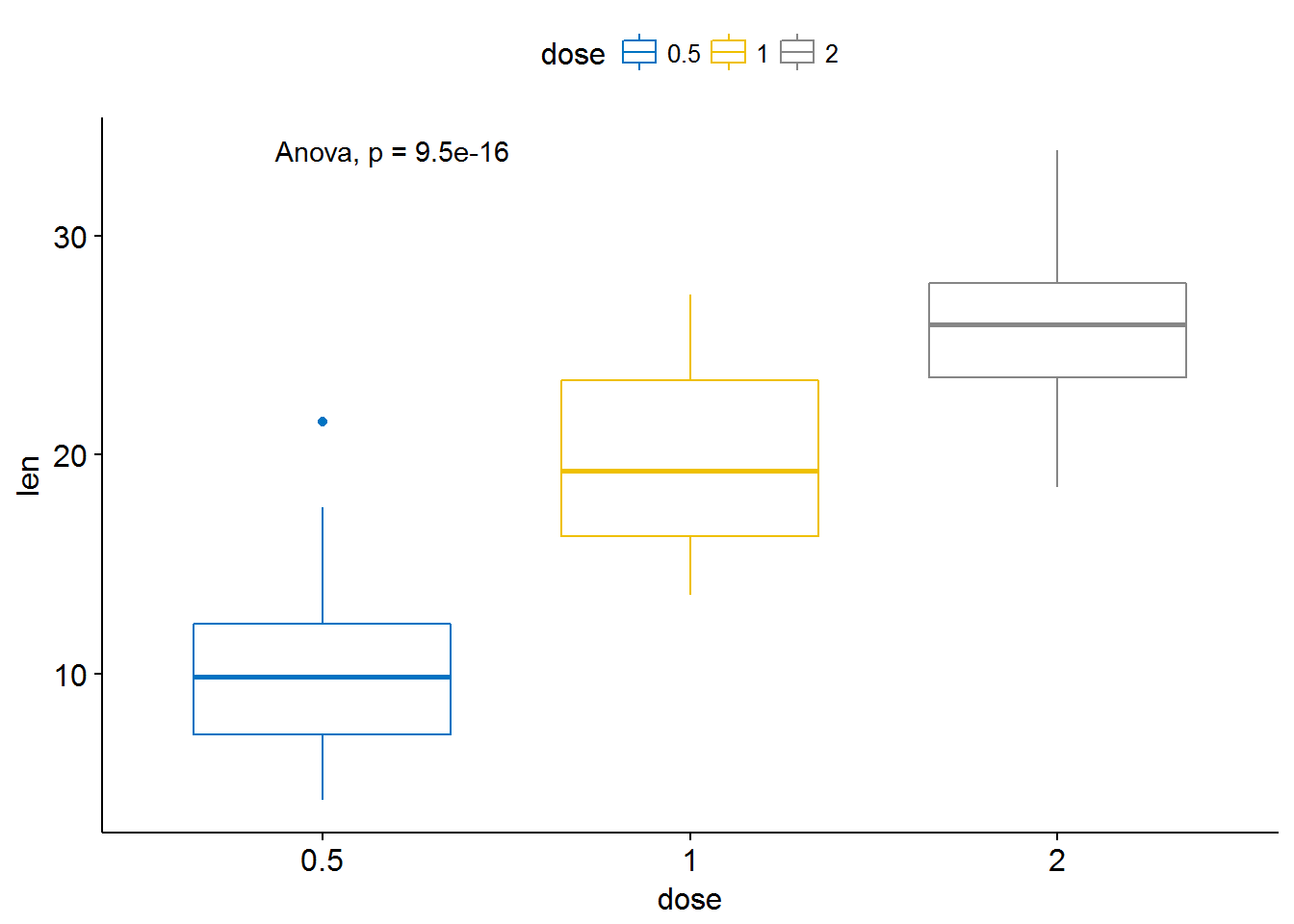
# Perorm pairwise comparisons
compare_means(len ~ dose, data = ToothGrowth)## # A tibble: 3 x 8
## .y. group1 group2 p p.adj p.format p.signif method
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
## 1 len 0.5 1 7.020855e-06 1.404171e-05 7.0e-06 **** Wilcoxon
## 2 len 0.5 2 8.406447e-08 2.521934e-07 8.4e-08 **** Wilcoxon
## 3 len 1 2 1.772382e-04 1.772382e-04 0.00018 *** Wilcoxon# Visualize: Specify the comparisons you want
my_comparisons <- list( c("0.5", "1"), c("1", "2"), c("0.5", "2") )
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
# stat_compare_means(comparisons = my_comparisons)+ # Add pairwise comparisons p-value
stat_compare_means(label.y = 50) # Add global 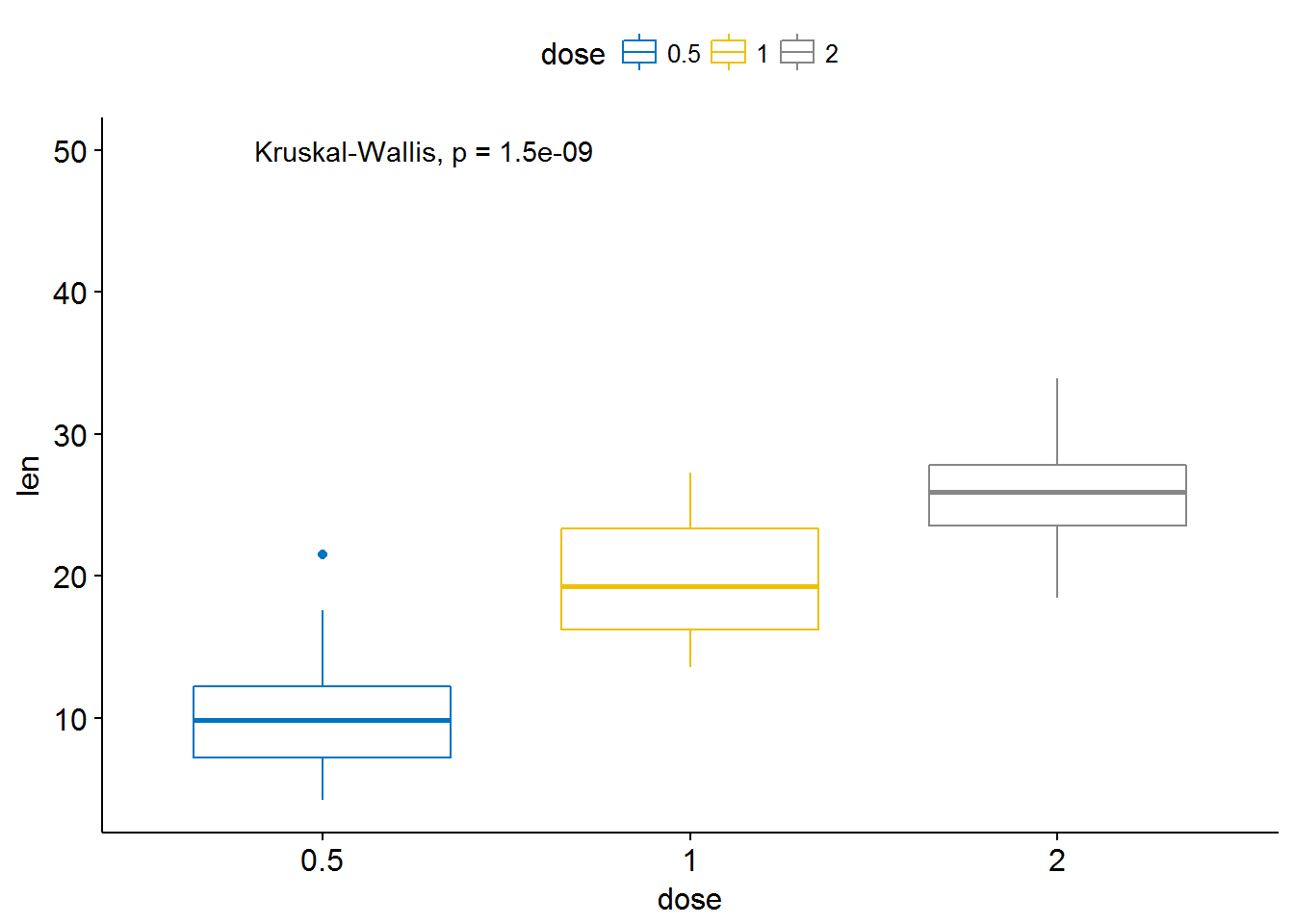
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
stat_compare_means(comparisons = my_comparisons, label.y = c(29, 35, 40))+
stat_compare_means(label.y = 45)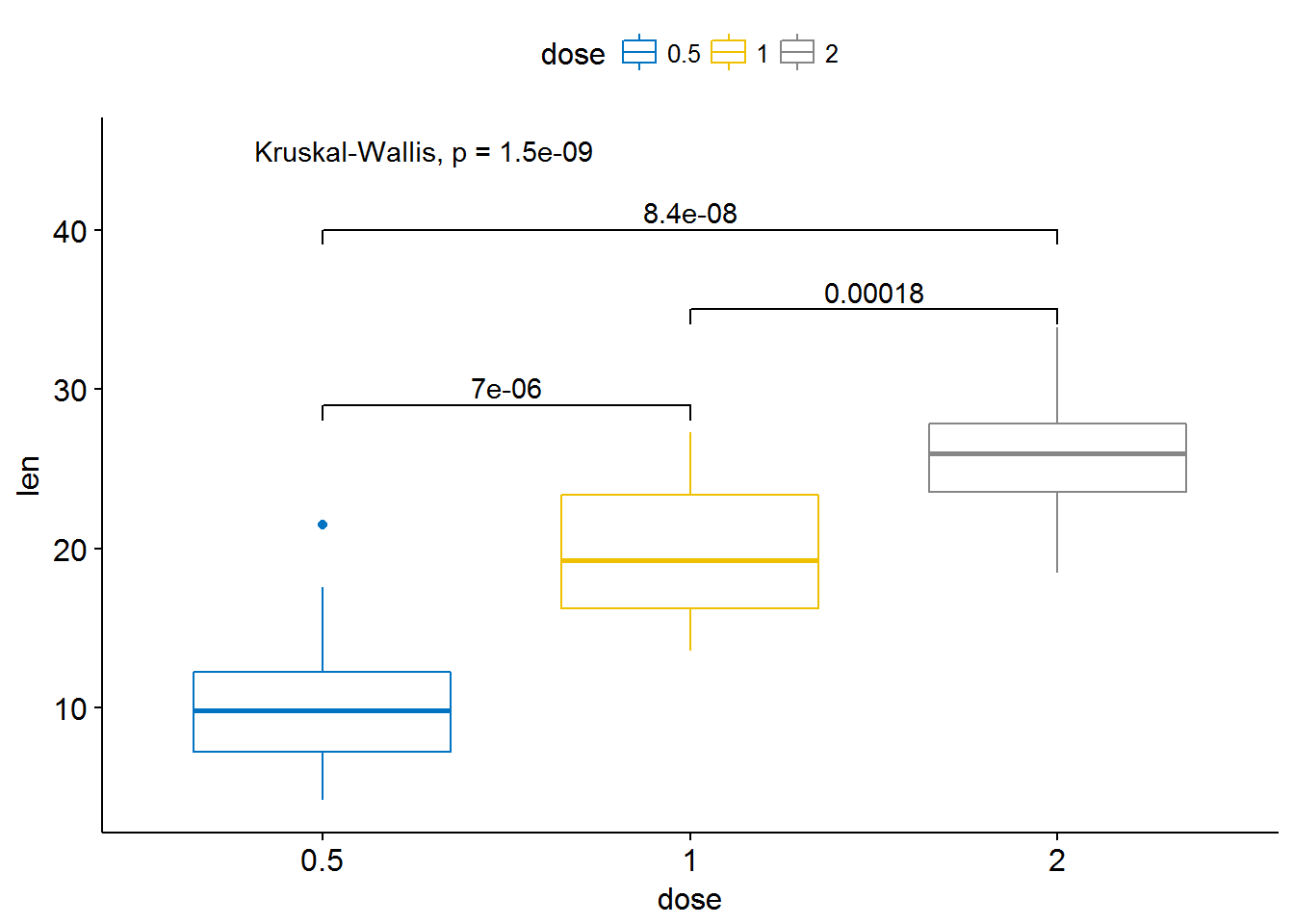
# Pairwise comparison against reference
compare_means(len ~ dose, data = ToothGrowth, ref.group = "0.5",
method = "t.test")## # A tibble: 2 x 8
## .y. group1 group2 p p.adj p.format p.signif method
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
## 1 len 0.5 1 6.697250e-09 6.697250e-09 6.7e-09 **** T-test
## 2 len 0.5 2 1.469534e-16 2.939068e-16 < 2e-16 **** T-test# Visualize
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
stat_compare_means(method = "anova", label.y = 40)+ # Add global p-value
stat_compare_means(label = "p.signif", method = "t.test",
ref.group = "0.5") # Pairwise comparison against reference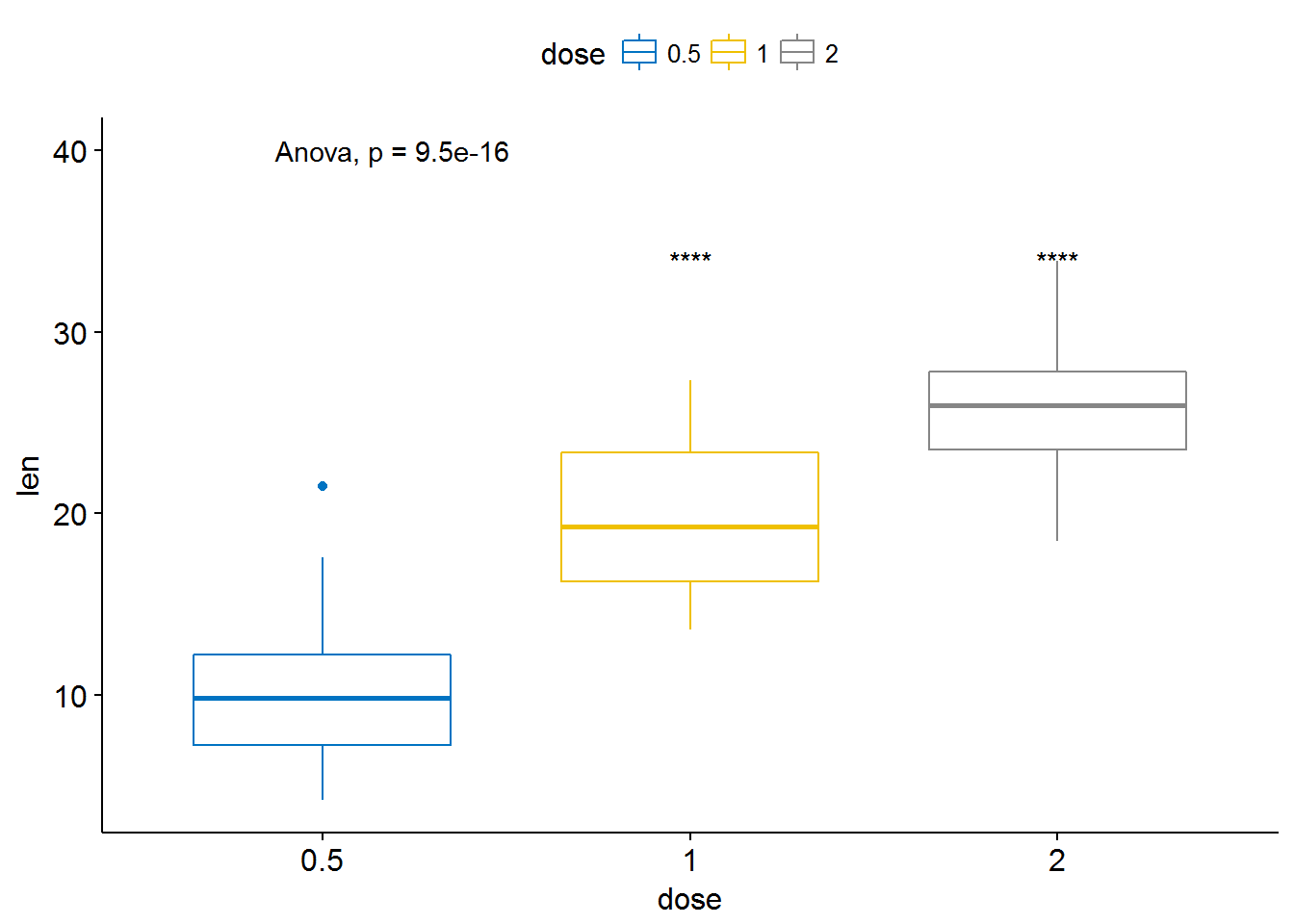
# Comparison of each group against base-mean
compare_means(len ~ dose, data = ToothGrowth, ref.group = ".all.",
method = "t.test")## # A tibble: 3 x 8
## .y. group1 group2 p p.adj p.format p.signif method
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
## 1 len .all. 0.5 1.244626e-06 3.733877e-06 1.2e-06 **** T-test
## 2 len .all. 1 5.667266e-01 5.667266e-01 0.57 ns T-test
## 3 len .all. 2 1.371704e-05 2.743408e-05 1.4e-05 **** T-test# Visualize
p + stat_compare_means( aes(label = ..p.signif..),
label.x = 1.5, label.y = 40)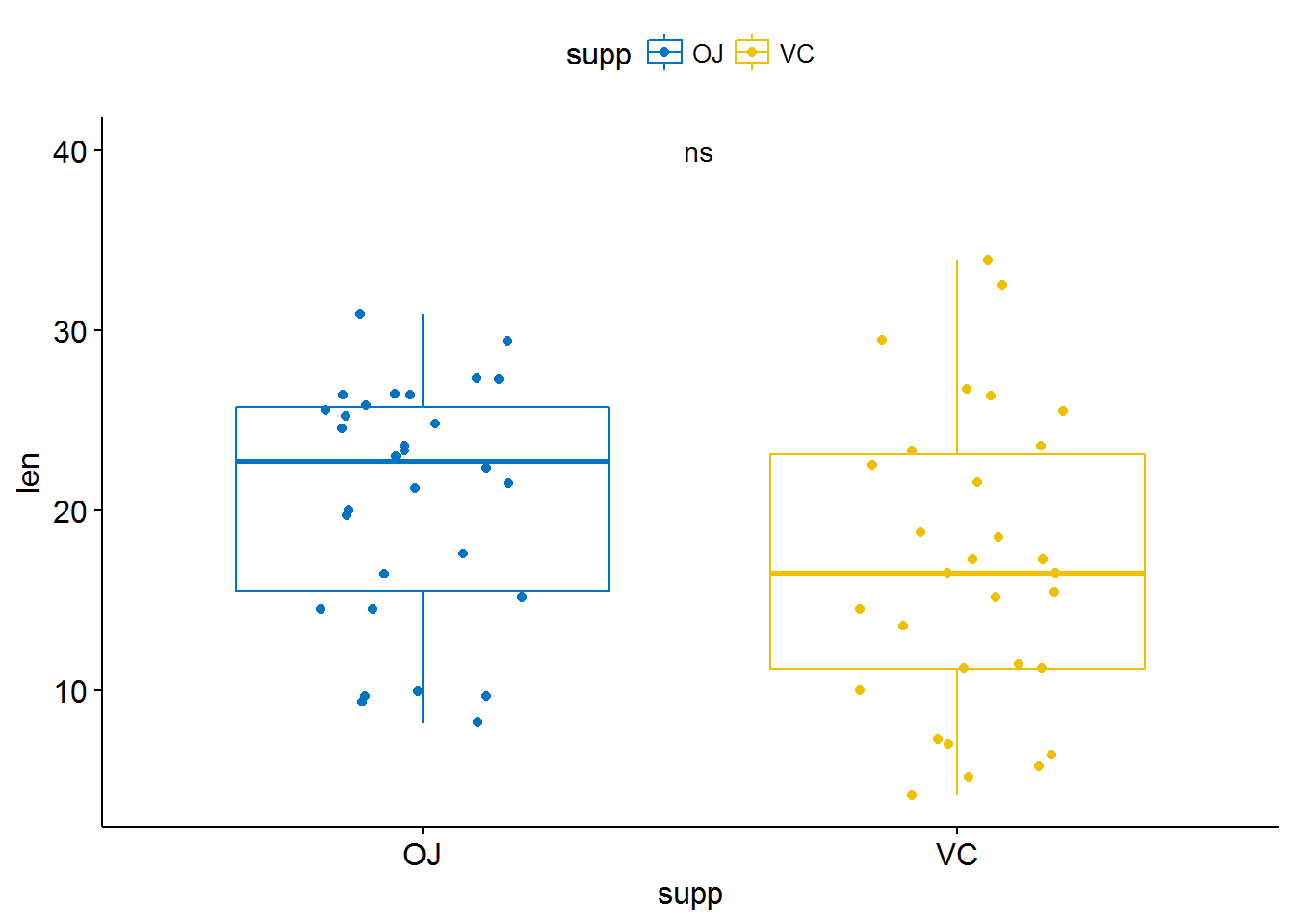
p + stat_compare_means( label = "p.signif", label.x = 1.5, label.y = 40)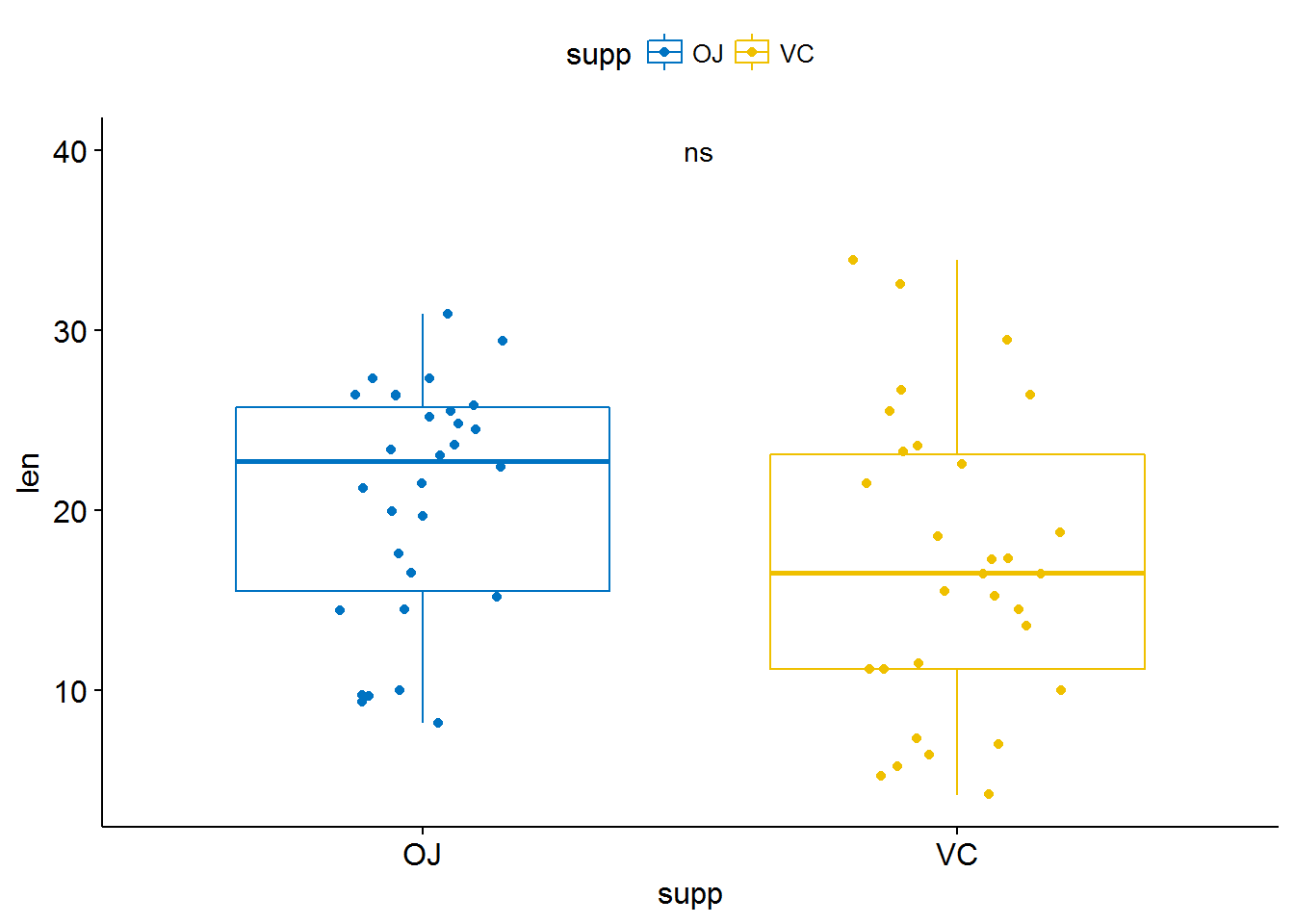
compare_means(len ~ supp, data = ToothGrowth, paired = TRUE)## # A tibble: 1 x 8
## .y. group1 group2 p p.adj p.format p.signif method
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr>
## 1 len OJ VC 0.004312554 0.004312554 0.0043 ** WilcoxonCompare two paired samples
ggpaired(ToothGrowth, x = "supp", y = "len",
color = "supp", line.color = "gray", line.size = 0.4,
palette = "jco")+
stat_compare_means(paired = TRUE)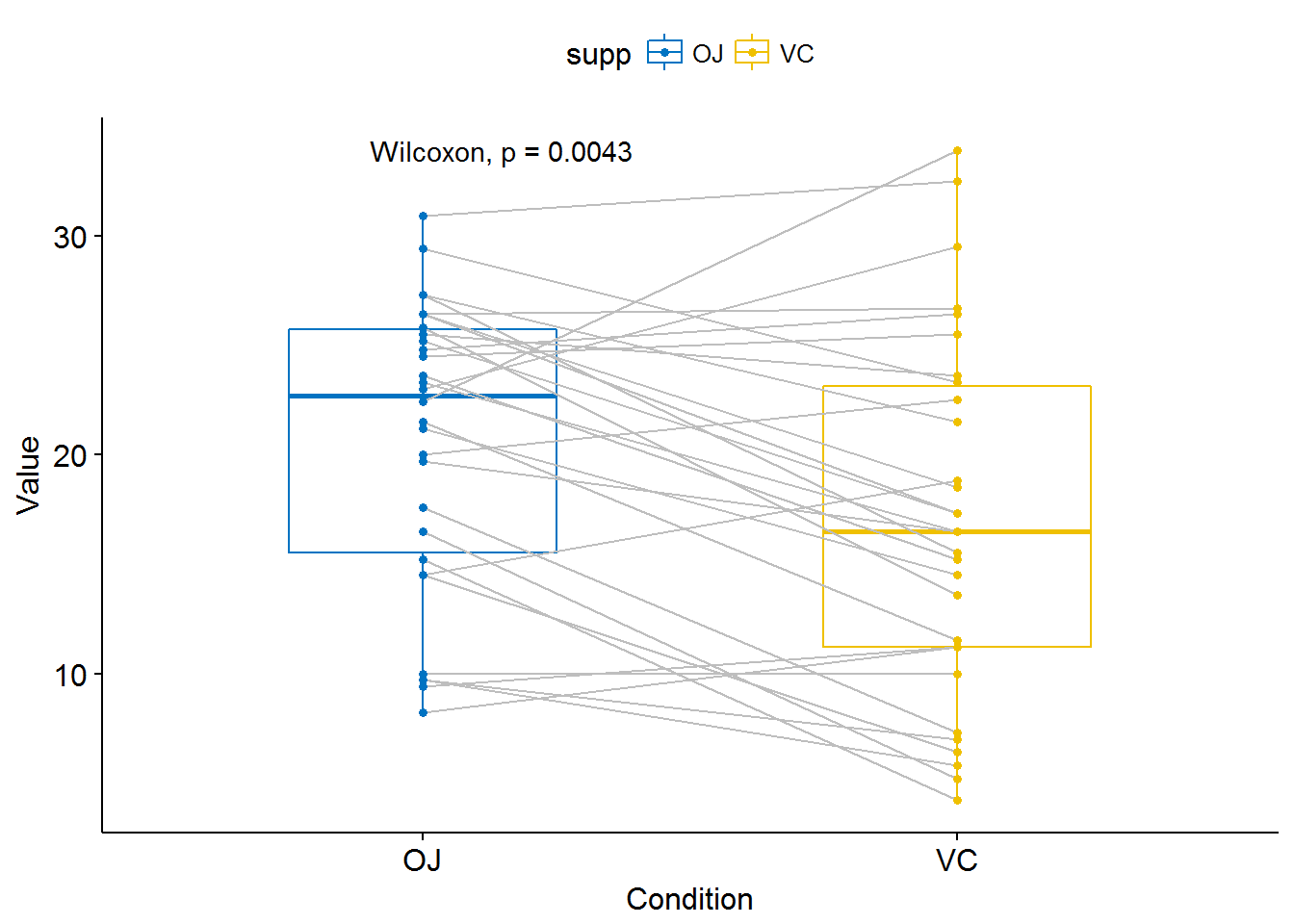
ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "dose", palette = "jco")+
stat_compare_means(method = "anova", label.y = 40)+ # Add global p-value
stat_compare_means(label = "p.signif", method = "t.test",
ref.group = ".all.") # Pairwise comparison against all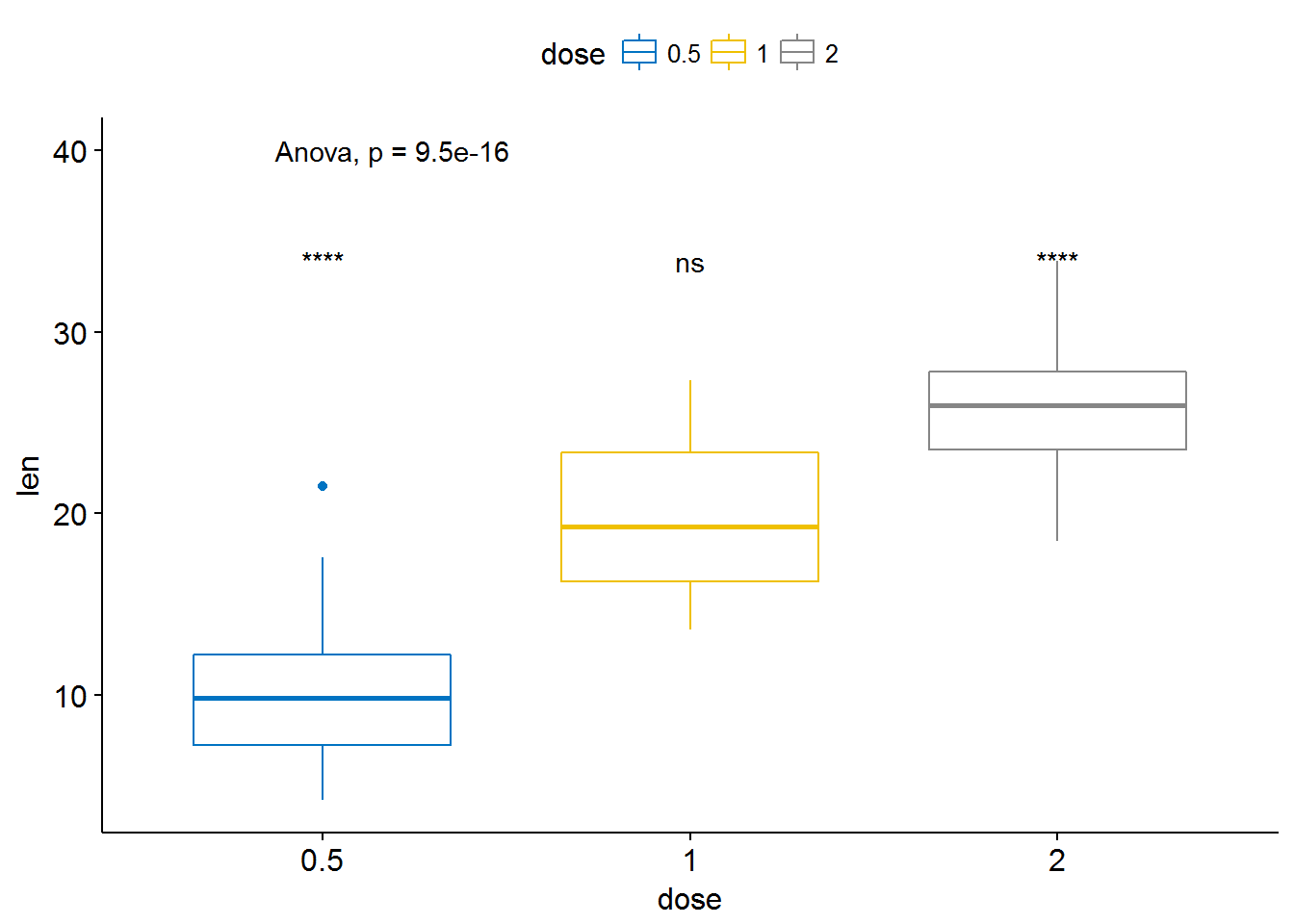
# Load myeloma data from survminer package
if(!require(survminer)) install.packages("survminer")## Loading required package: survminer## Warning: package 'survminer' was built under R version 3.3.3data("myeloma", package = "survminer")
# Perform the test
compare_means(DEPDC1 ~ molecular_group, data = myeloma,
ref.group = ".all.", method = "t.test")## # A tibble: 7 x 8
## .y. group1 group2 p p.adj p.format
## <chr> <chr> <chr> <dbl> <dbl> <chr>
## 1 DEPDC1 .all. Cyclin D-1 1.496896e-01 4.490687e-01 0.14969
## 2 DEPDC1 .all. Cyclin D-2 5.231428e-01 1.000000e+00 0.52314
## 3 DEPDC1 .all. Hyperdiploid 2.815333e-04 1.689200e-03 0.00028
## 4 DEPDC1 .all. Low bone disease 5.083985e-03 2.541992e-02 0.00508
## 5 DEPDC1 .all. MAF 8.610664e-02 3.444265e-01 0.08611
## 6 DEPDC1 .all. MMSET 5.762908e-01 1.000000e+00 0.57629
## 7 DEPDC1 .all. Proliferation 1.241416e-09 8.689910e-09 1.2e-09
## # ... with 2 more variables: p.signif <chr>, method <chr># Visualize the expression profile
ggboxplot(myeloma, x = "molecular_group", y = "DEPDC1", color = "molecular_group",
add = "jitter", legend = "none") +
rotate_x_text(angle = 45)+
geom_hline(yintercept = mean(myeloma$DEPDC1), linetype = 2)+ # Add horizontal line at base mean
stat_compare_means(method = "anova", label.y = 1600)+ # Add global annova p-value
stat_compare_means(label = "p.signif", method = "t.test",
ref.group = ".all.") # Pairwise comparison against all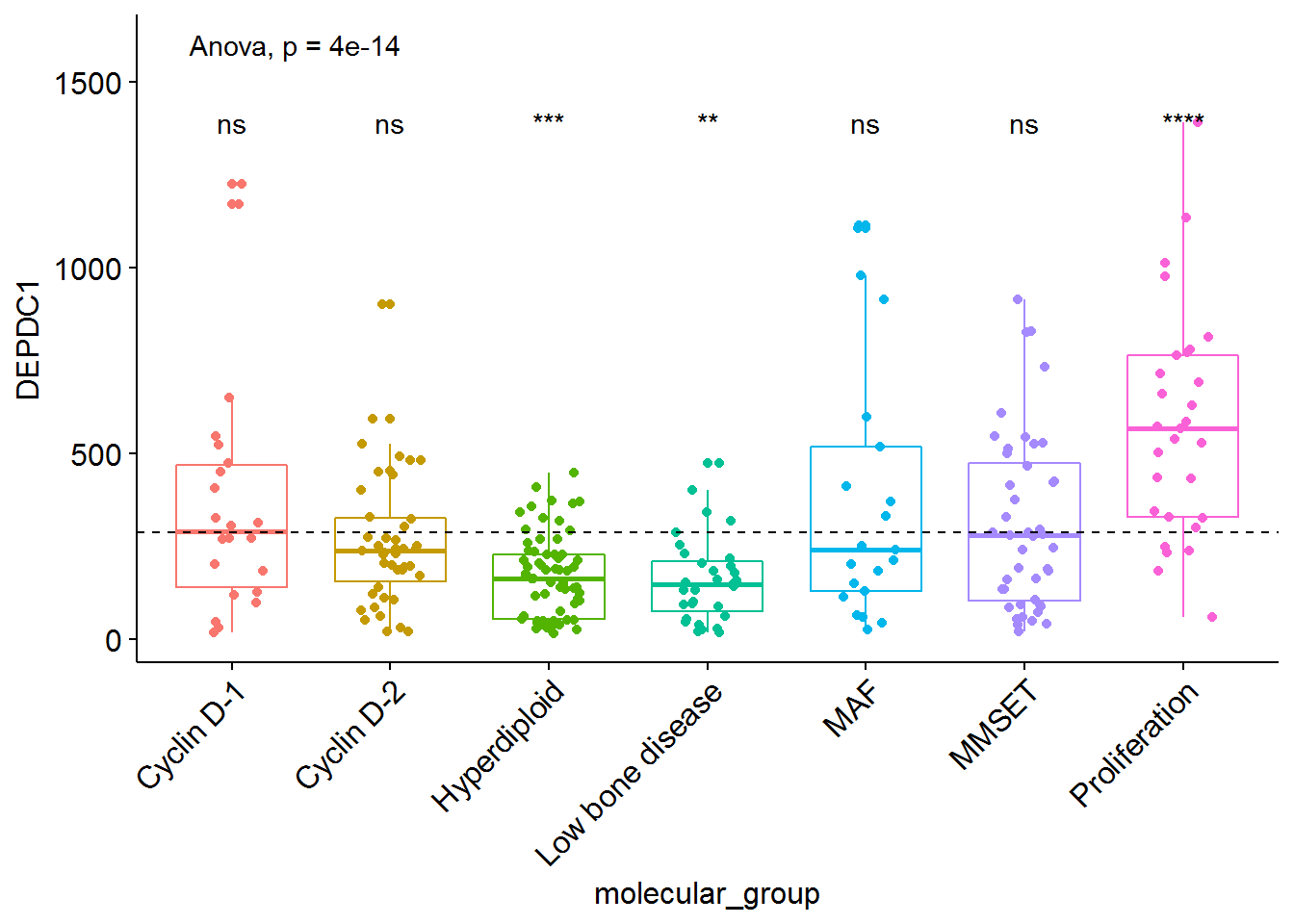
# Visualize the expression profile
ggboxplot(myeloma, x = "molecular_group", y = "DEPDC1", color = "molecular_group",
add = "jitter", legend = "none") +
rotate_x_text(angle = 45)+
geom_hline(yintercept = mean(myeloma$DEPDC1), linetype = 2)+ # Add horizontal line at base mean
stat_compare_means(method = "anova", label.y = 1600)+ # Add global annova p-value
stat_compare_means(label = "p.signif", method = "t.test",
ref.group = ".all.", hide.ns = TRUE) # Pairwise comparison against all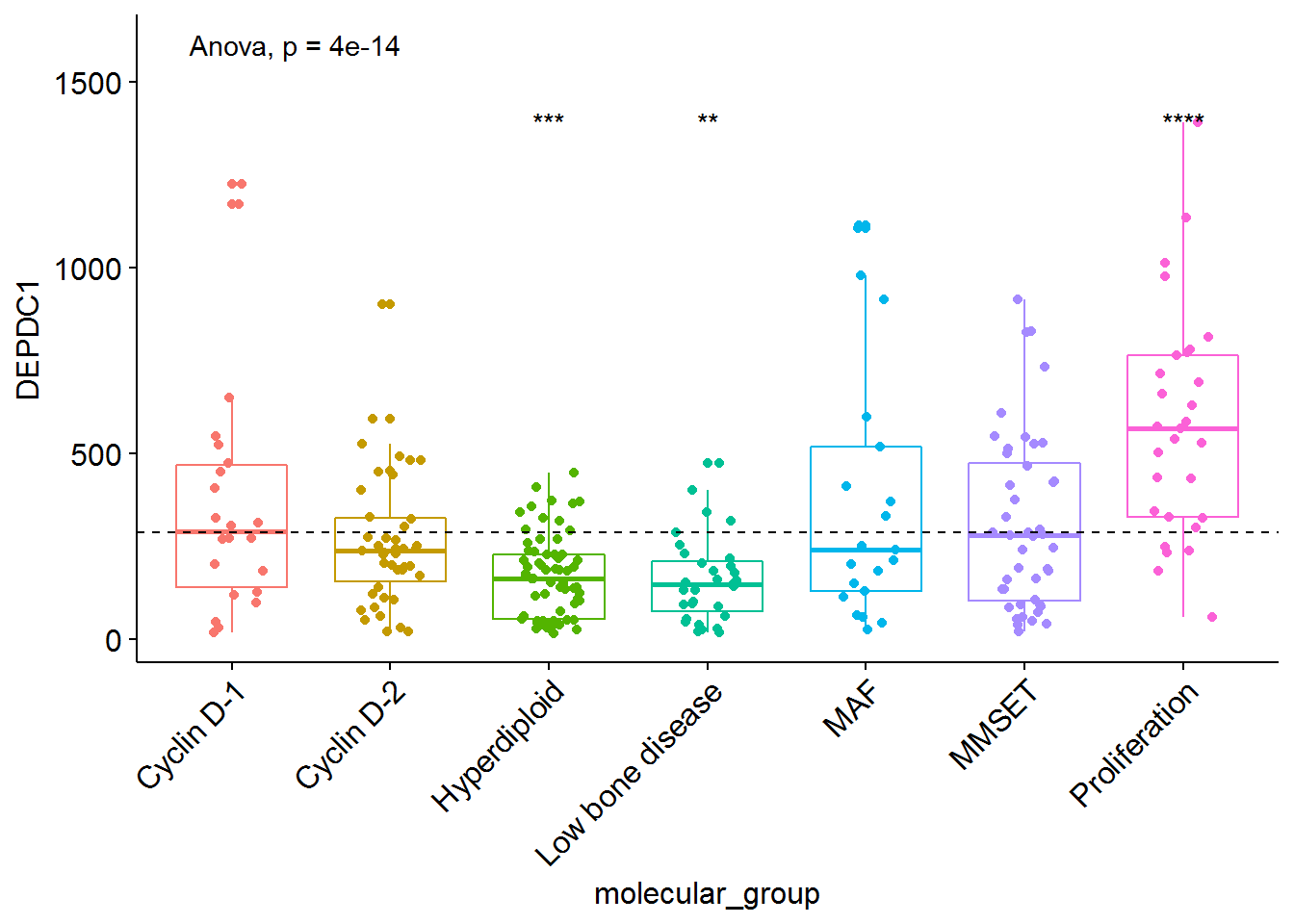
compare_means(len ~ supp, data = ToothGrowth,
group.by = "dose")## # A tibble: 3 x 9
## dose .y. group1 group2 p p.adj p.format p.signif
## <dbl> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr>
## 1 0.5 len OJ VC 0.023186427 0.04637285 0.023 *
## 2 1.0 len OJ VC 0.004030367 0.01209110 0.004 **
## 3 2.0 len OJ VC 1.000000000 1.00000000 1.000 ns
## # ... with 1 more variables: method <chr># Box plot facetted by "dose"
p <- ggboxplot(ToothGrowth, x = "supp", y = "len",
color = "supp", palette = "jco",
add = "jitter",
facet.by = "dose", short.panel.labs = FALSE)
# Use only p.format as label. Remove method name.
p + stat_compare_means(label = "p.format")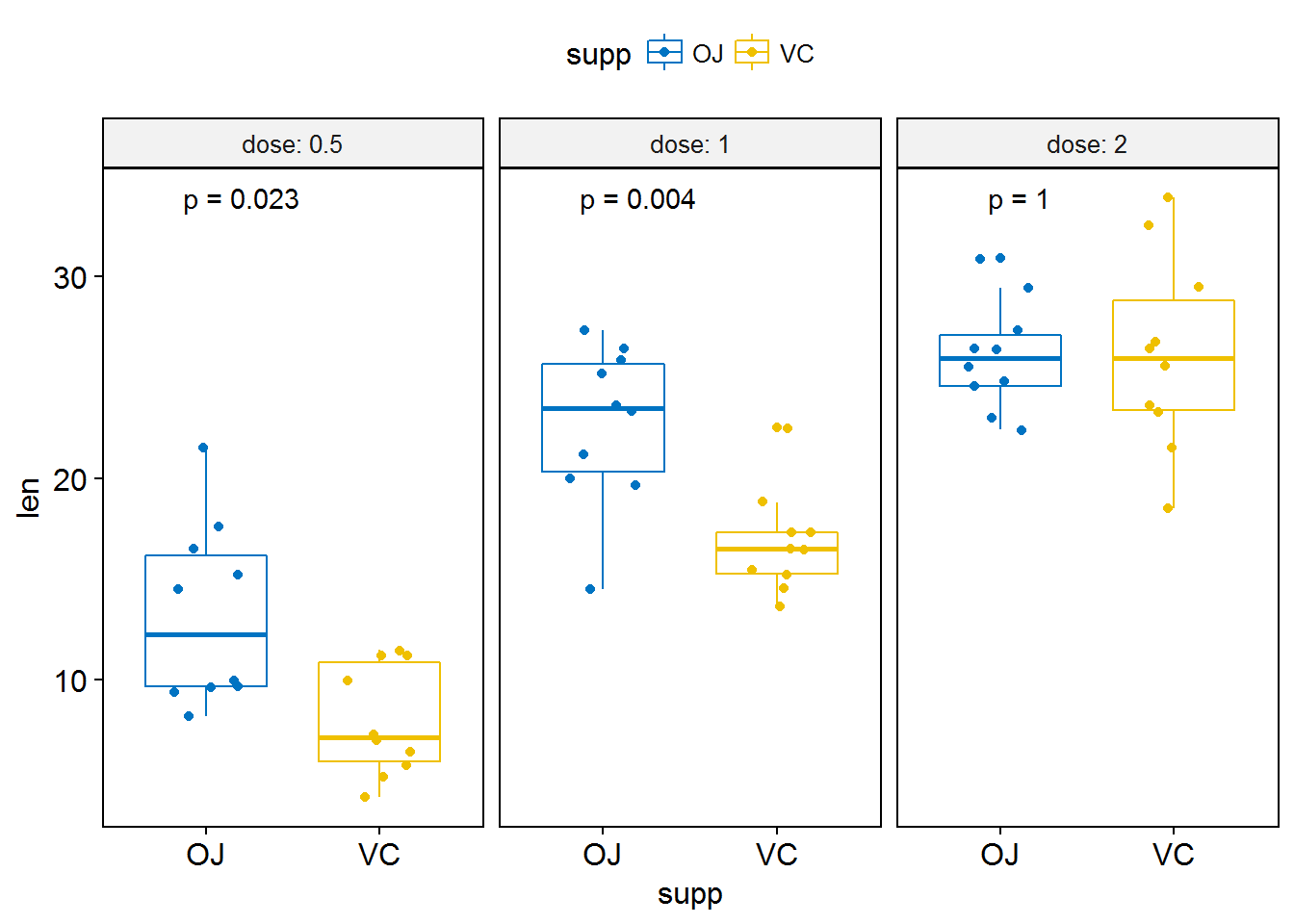
# Or use significance symbol as label
p + stat_compare_means(label = "p.signif", label.x = 1.5)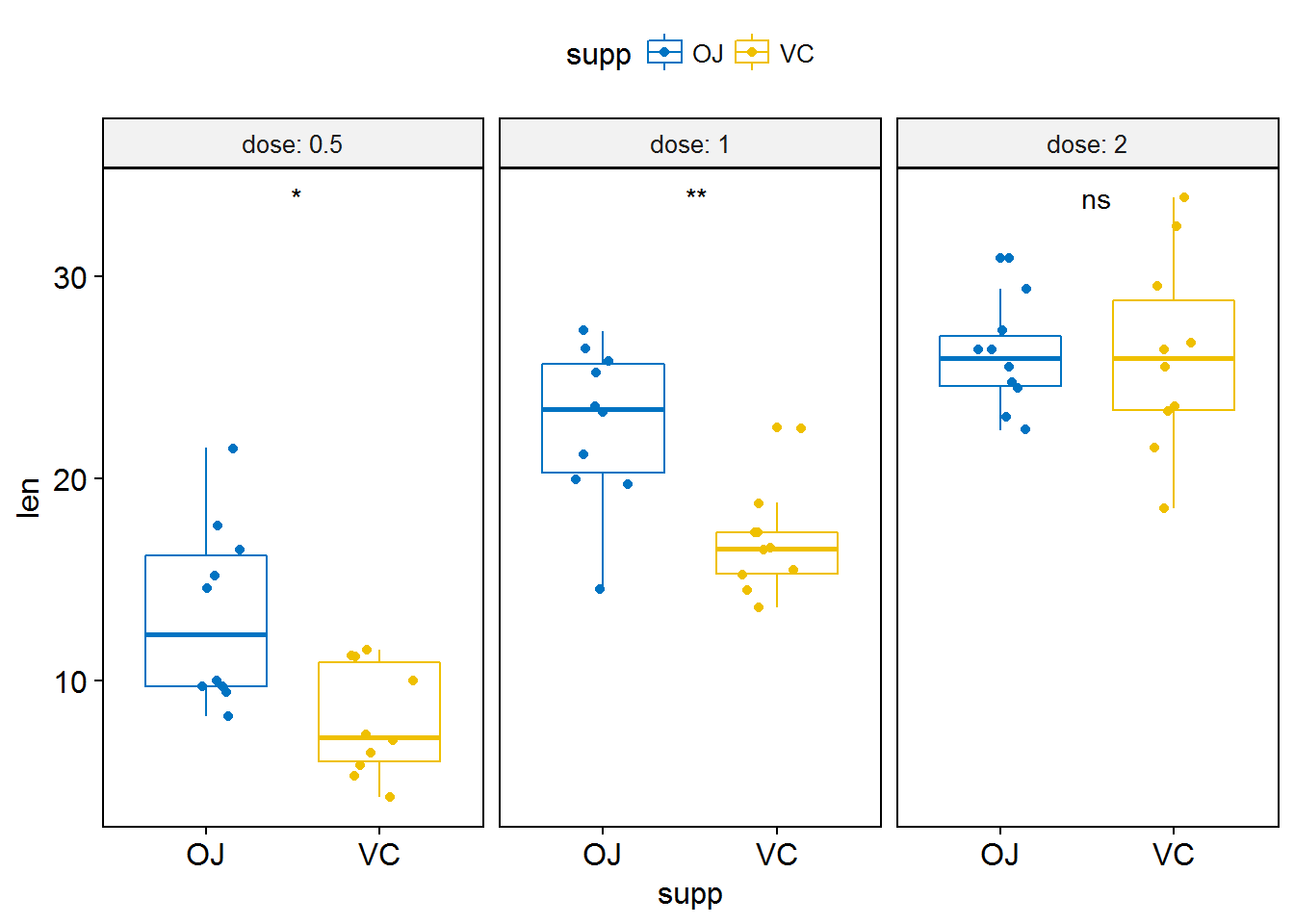
p <- ggboxplot(ToothGrowth, x = "dose", y = "len",
color = "supp", palette = "jco",
add = "jitter")
p + stat_compare_means(aes(group = supp))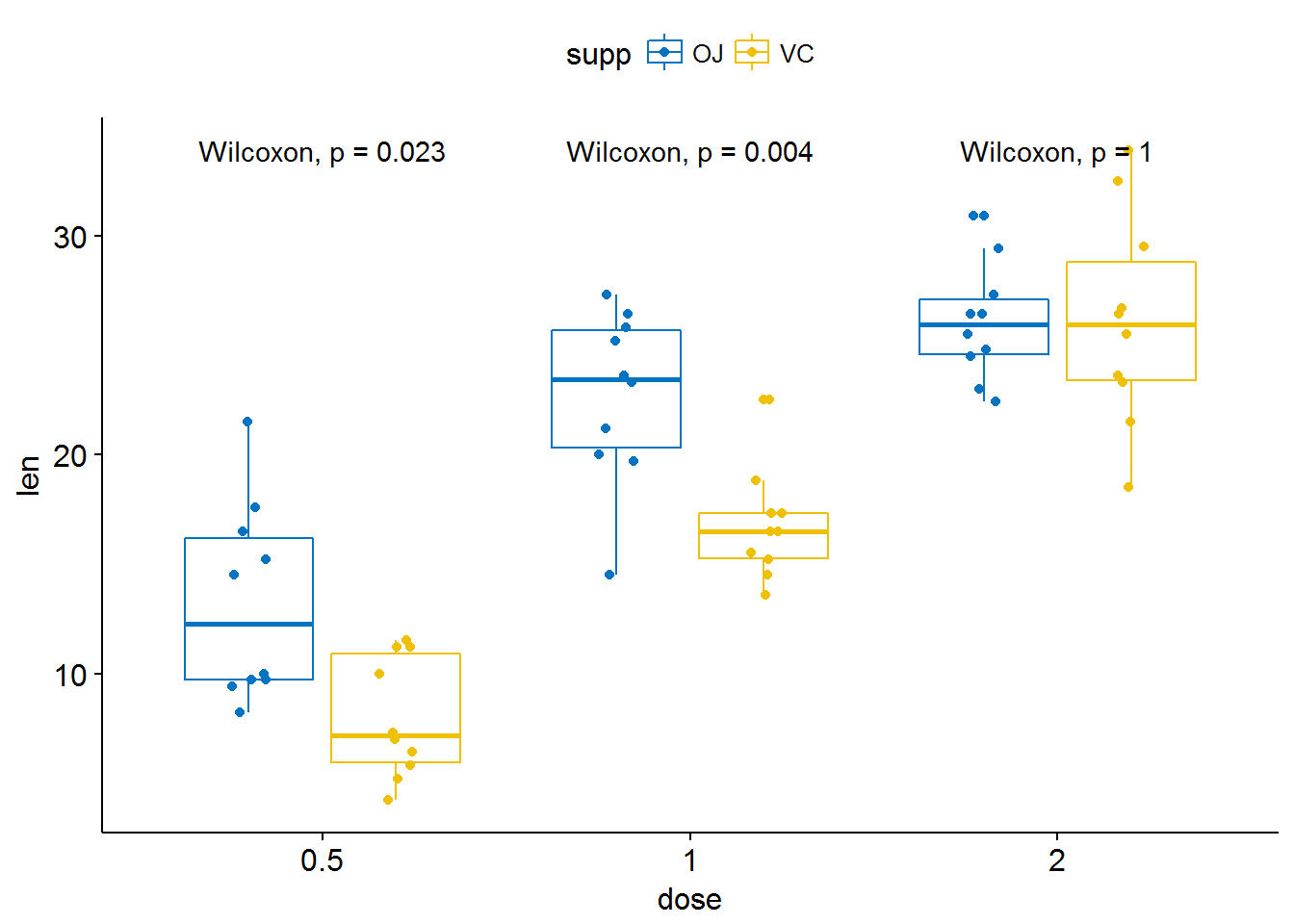
# Show only p-value
p + stat_compare_means(aes(group = supp), label = "p.format")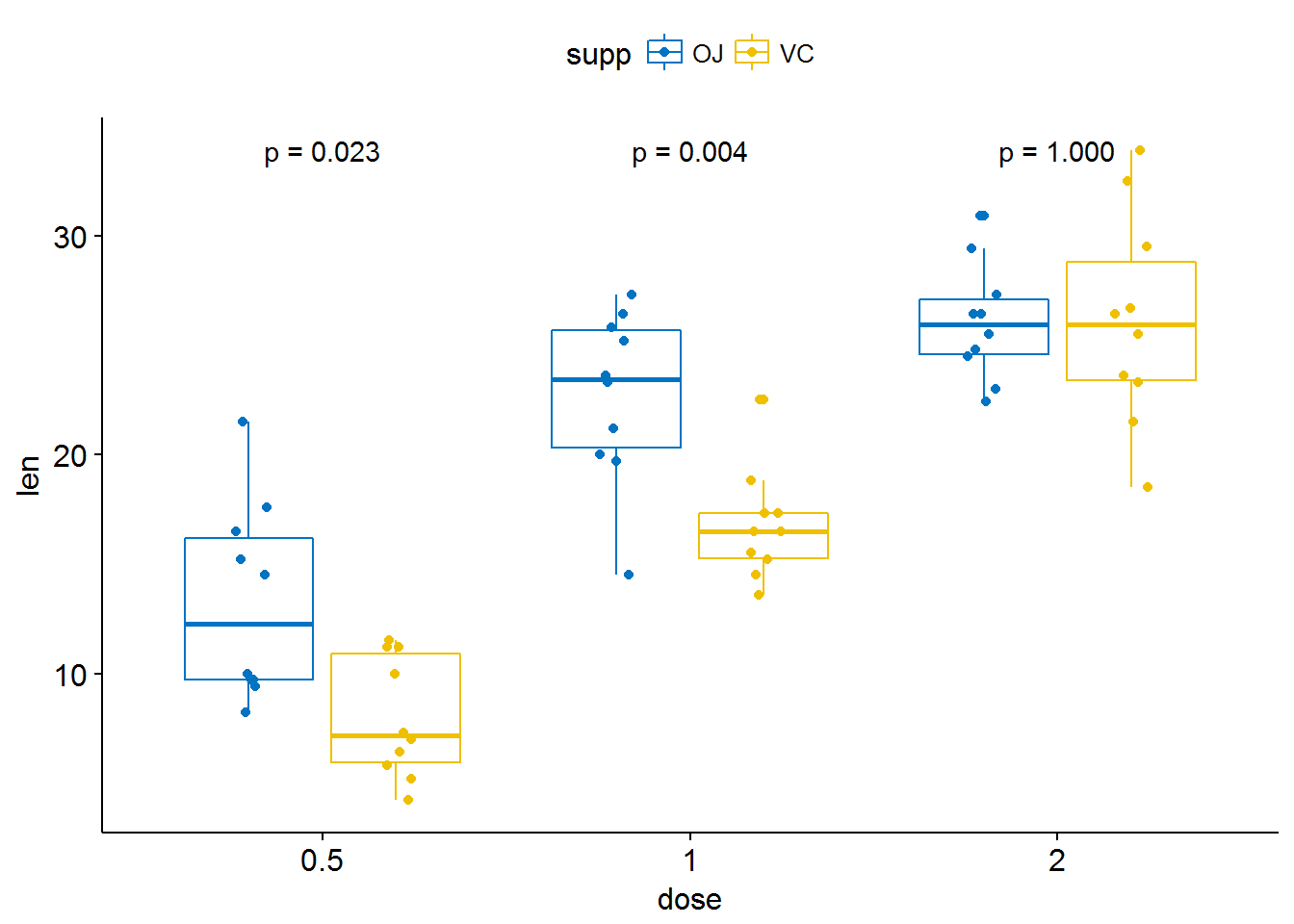
# Use significance symbol as label
p + stat_compare_means(aes(group = supp), label = "p.signif")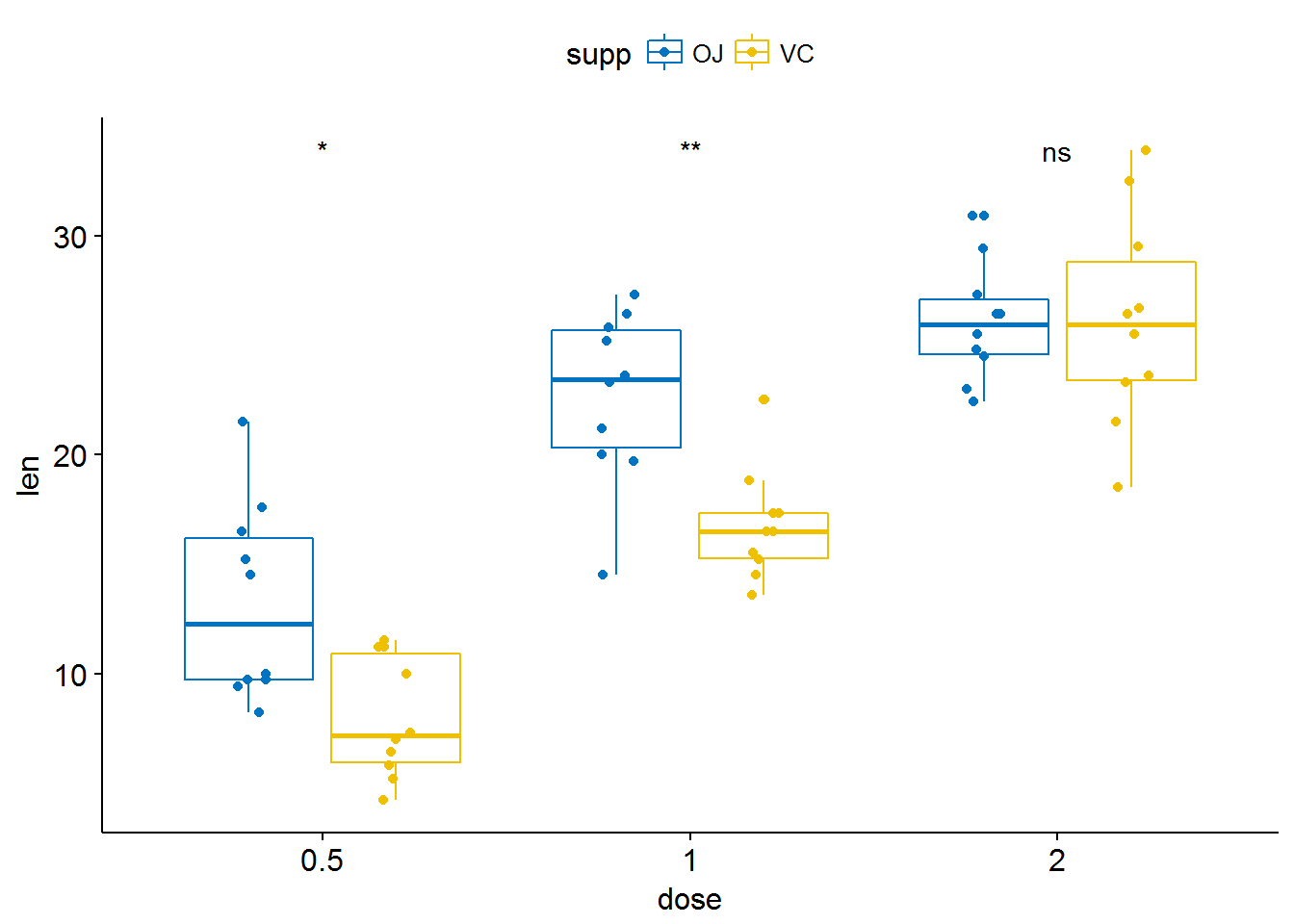
compare_means(len ~ supp, data = ToothGrowth,
group.by = "dose", paired = TRUE)## # A tibble: 3 x 9
## dose .y. group1 group2 p p.adj p.format p.signif
## <dbl> <chr> <chr> <chr> <dbl> <dbl> <chr> <chr>
## 1 0.5 len OJ VC 0.03296938 0.06593876 0.033 *
## 2 1.0 len OJ VC 0.01905889 0.05717667 0.019 *
## 3 2.0 len OJ VC 1.00000000 1.00000000 1.000 ns
## # ... with 1 more variables: method <chr># Box plot facetted by "dose"
p <- ggpaired(ToothGrowth, x = "supp", y = "len",
color = "supp", palette = "jco",
line.color = "gray", line.size = 0.4,
facet.by = "dose", short.panel.labs = FALSE)
# Use only p.format as label. Remove method name.
p + stat_compare_means(label = "p.format", paired = TRUE)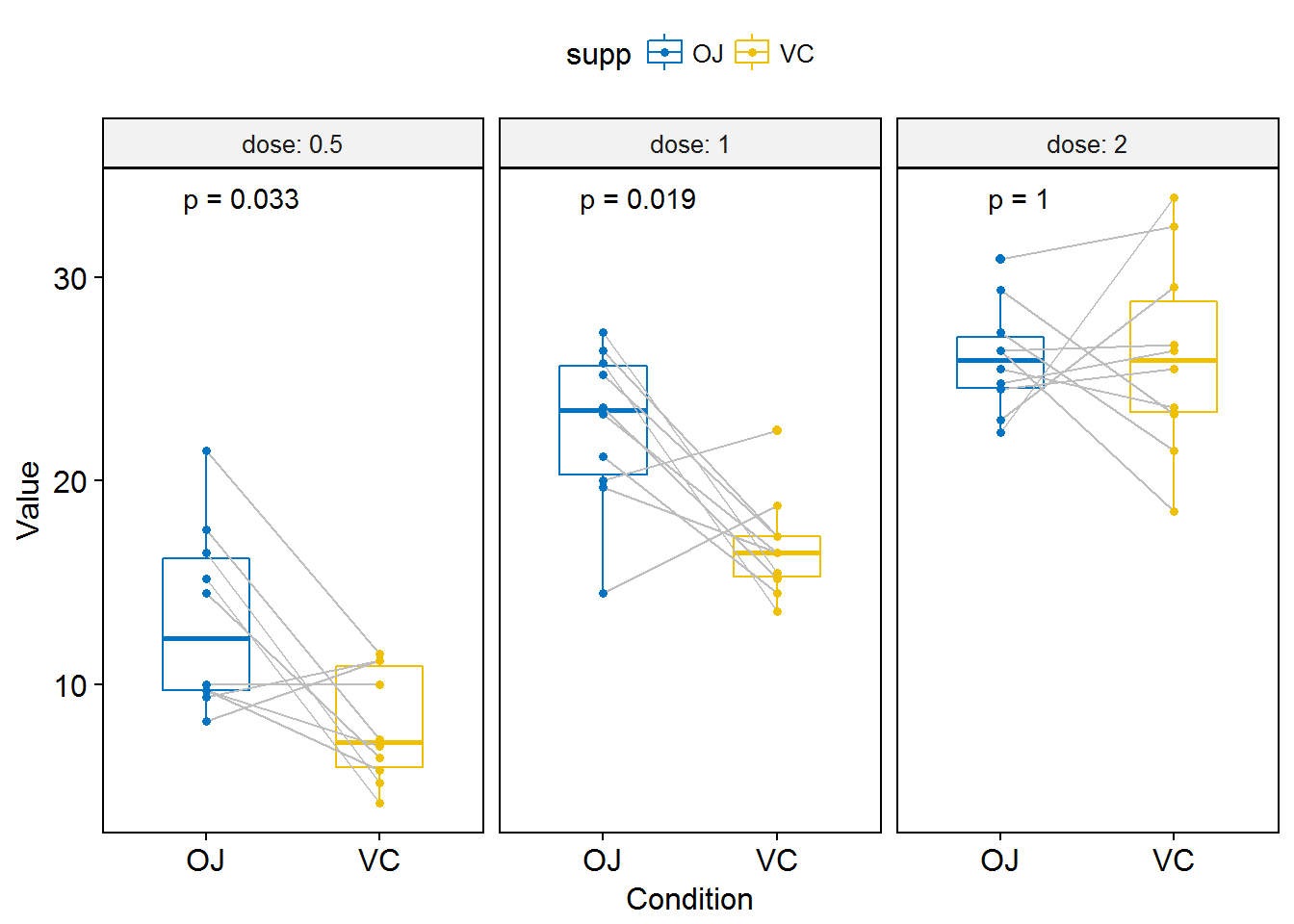
# Bar plot of mean +/-se
ggbarplot(ToothGrowth, x = "dose", y = "len", add = "mean_se")+
stat_compare_means() + # Global p-value
stat_compare_means(ref.group = "0.5", label = "p.signif",
label.y = c(22, 29)) # compare to ref.group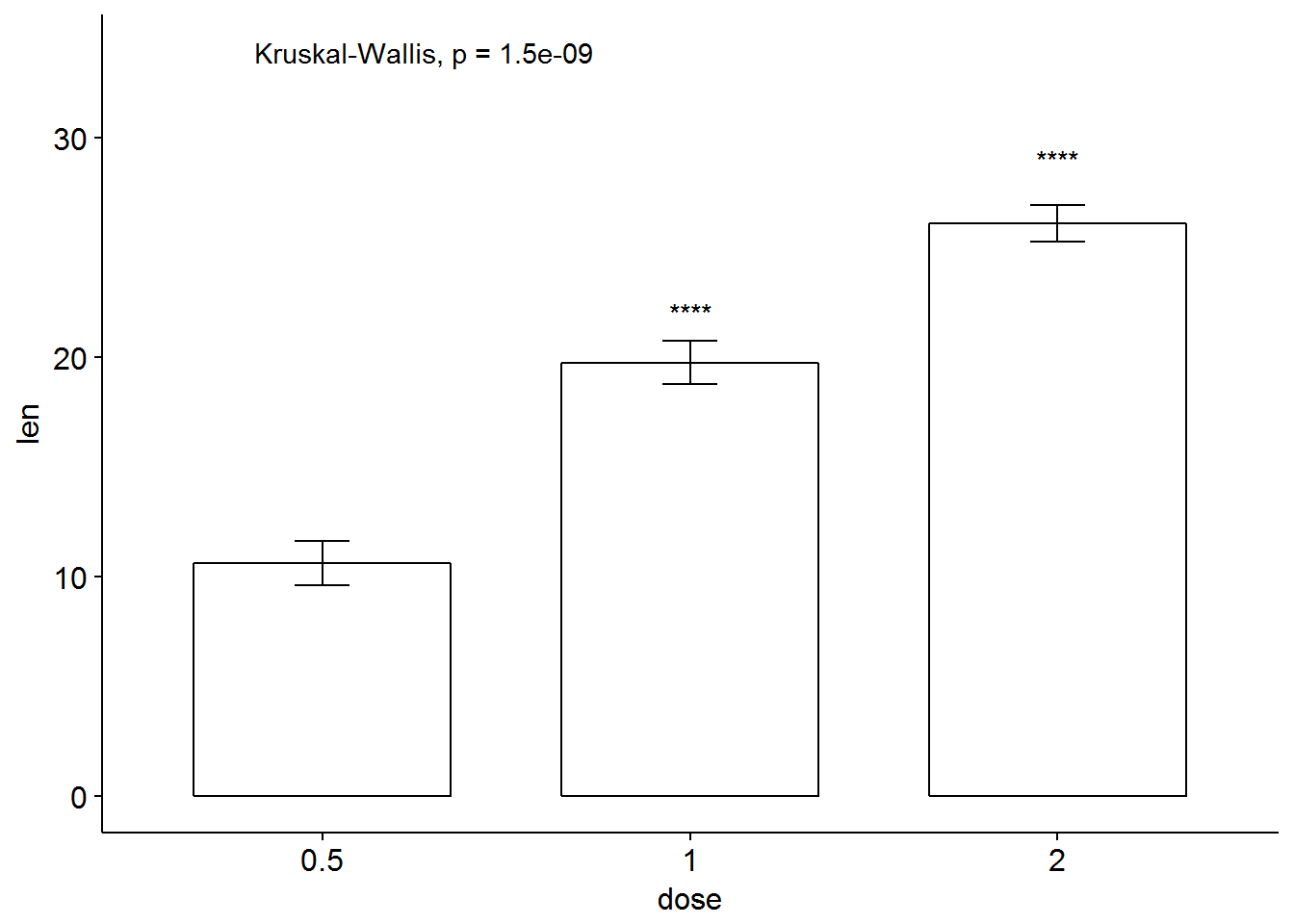
# Line plot of mean +/-se
ggline(ToothGrowth, x = "dose", y = "len", add = "mean_se")+
stat_compare_means() + # Global p-value
stat_compare_means(ref.group = "0.5", label = "p.signif",
label.y = c(22, 29)) 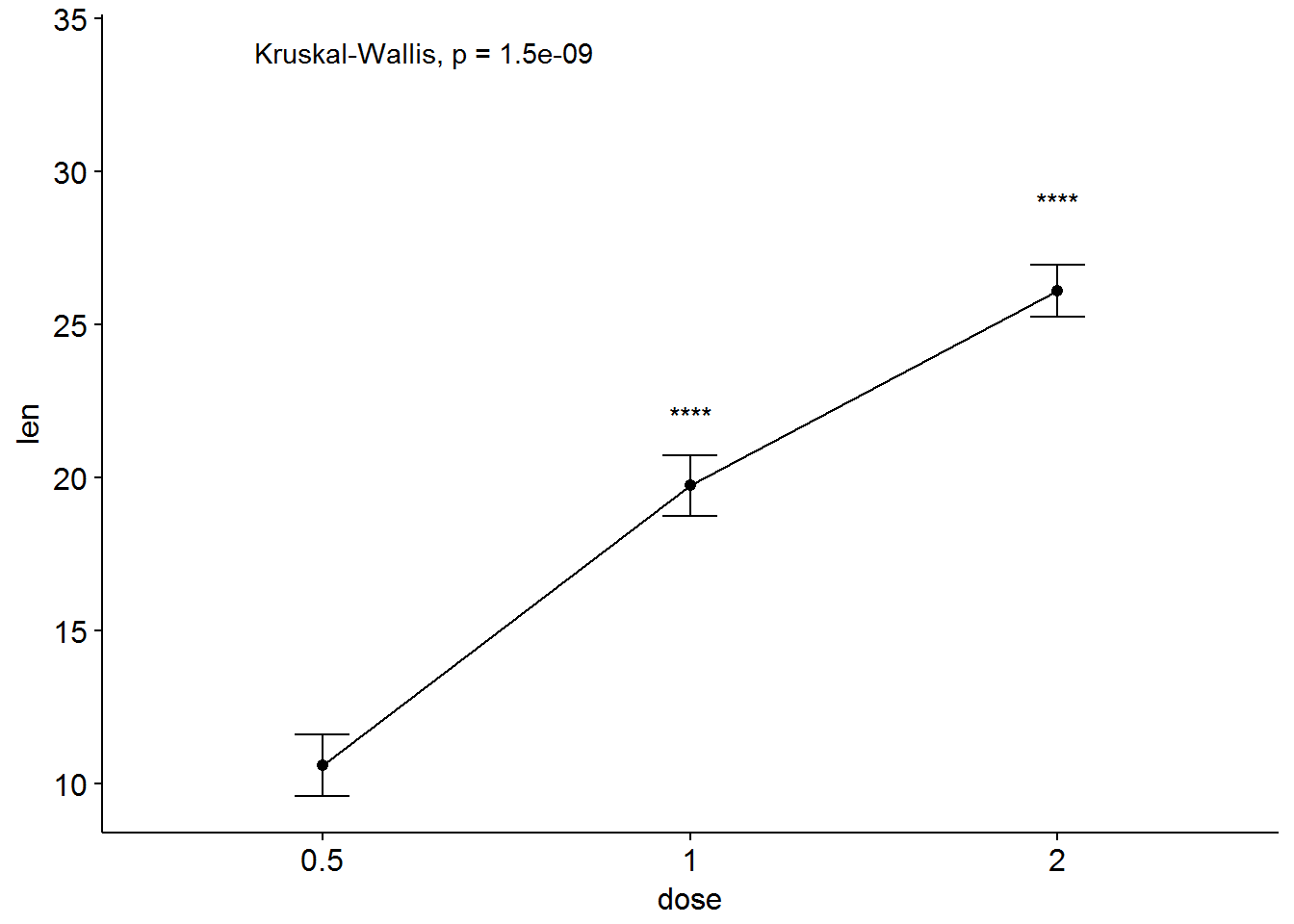
ggbarplot(ToothGrowth, x = "dose", y = "len", add = "mean_se",
color = "supp", palette = "jco",
position = position_dodge(0.8))+
stat_compare_means(aes(group = supp), label = "p.signif", label.y = 29)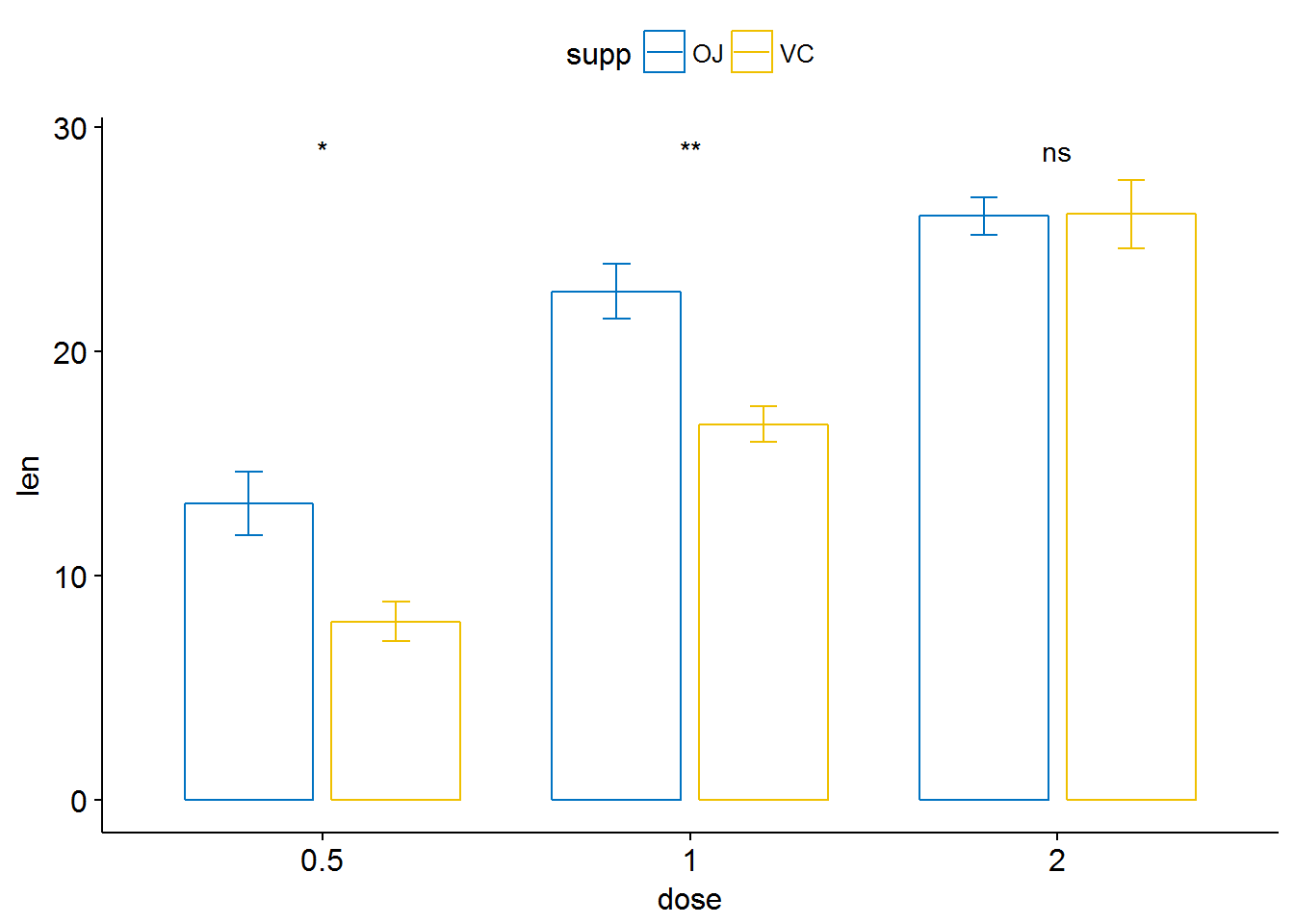
#
ggline(ToothGrowth, x = "dose", y = "len", add = "mean_se",
color = "supp", palette = "jco")+
stat_compare_means(aes(group = supp), label = "p.signif",
label.y = c(16, 25, 29))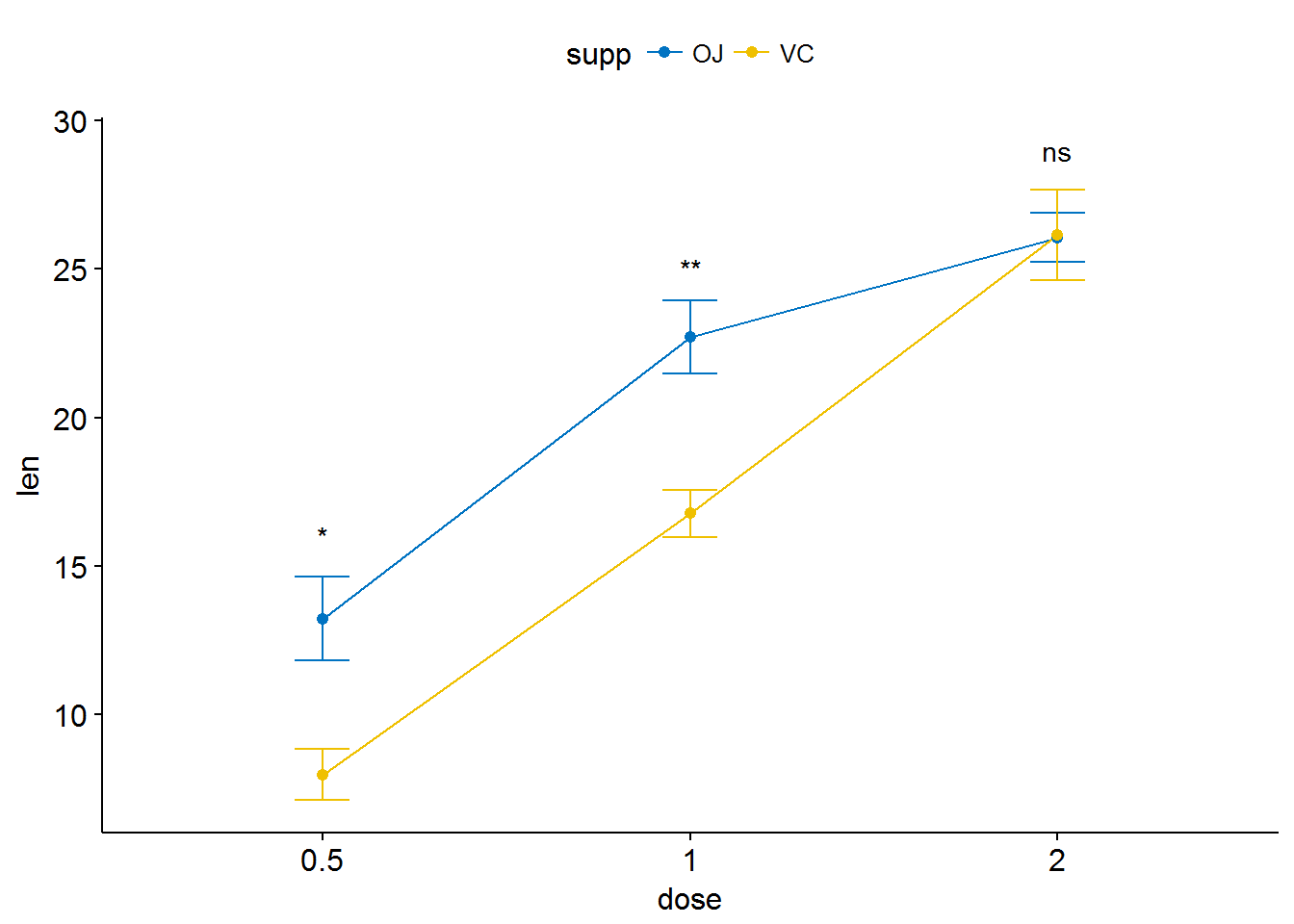
Reference: https://www.r-bloggers.com/add-p-values-and-significance-levels-to-ggplots/
Share this post
Twitter
Google+
Facebook
Reddit
LinkedIn
StumbleUpon
Email